- 事例No.PC-TRNX254496
-
 参考価格 (税込):
参考価格 (税込):
1,156,100円事例追加日:2026/2/6Gaussian・LAMMPS・GROMACS 併用向けワークステーション
用途:Gaussian16、LAMMPS、GROMACSお客さまからのご相談内容
反応機構解析、タンパク質ドッキング、インシリコ (計算化学) スクリーニングを想定した計算用PCを導入したい。 現在はGaussian16 を中心に、LAMMPSとGROMACS も併用しているが、計算負荷が増え、現行PCでは性能不足が顕著になっている。 将来的にはLinux系のOSに変更し、LAMMPSとGROMACSをメインにすることも視野に入れている。 予算は約100万円 (超過も可)。 想…テガラからのご提案
CPUとメモリについて Gaussian は計算の多くを CPU で処理するため、並列利用するコア数と実効クロックが計算時間に直結します。 今回は、ご予算内で並列計算時の性能を確保できる、Ryzen 9 9950X (16コア) を採用しました。 メモリはご要望に合わせて128GB (32GB×4枚) を搭載しています。 Ryzen 9 9950X は最大 192GB まで対応しますが、今回の構成…主な仕様
CPU AMD Ryzen9 9950X 4.30GHz (Boost時最大5.70GHz) 16C/32T メモリ 合計128GB DDR5 5600 32GB x 4 ストレージ1 2TB SSD M.2 NVMe Gen4 ビデオ NVIDIA GeForce RTX5090 32GB ネットワーク on board (2.5G x1) Wi-Fi,Bluetooth 筐体+電源 ミドルタワー型筐体+1500W 80PLUS TITANIUM OS Microsoft Windows 11 Professional 64bit
- 事例No.PC-TE1D265386
-
 参考価格 (税込):
参考価格 (税込):
2,629,000円事例追加日:2026/2/5分子動力学計算向けワークステーション
用途:ナノテク・材料研究における分子動力学計算お客さまからのご相談内容
分子動力学計算に使用するワークステーションの見積もりを希望する。 AMD EPYC 9634 を用いた大規模並列計算を想定し、ECCメモリ 256GBを搭載したい。 年度末までの納入できる構成であることが条件なので、パーツの入手性にも配慮して欲しい。テガラからのご提案
分子動力学計算はCPU性能とメモリ帯域の影響が大きいため、処理負荷に対して安定した計算を行える構成を中心に検討しました。 お客さまの希望スペックを確認したうえで、現行世代の入手性や長期運用を考慮し、同等クラスのパーツを組み合わせたワークステーションをご提案しています。 CPUについて ご相談時点ではEPYC 9634の入手性が芳しくなかったため、現行世代のCPUから計算効率と入手性のバランスを踏ま…主な仕様
CPU AMD EPYC 9655 2.60GHz 96C/192T メモリ 合計256GB DDR5 6400 REG ECC 32GB x 8 ストレージ 1TB SSD M.2 NVMe Gen5 ビデオ NVIDIA RTX PRO2000 16GB (MiniDisplayPort x4) ネットワーク on board (10GBase-T x2) 筐体+電源 ミドルタワー筐体 1000W 80PLUS PLATINUM OS Ubuntu 24.04
- 事例No.PC-TUNS254264
-
 参考価格 (税込):
参考価格 (税込):
331,100円事例追加日:2025/12/19HSPiP対応ワークステーション (推奨構成)
用途:HSPiPお客さまからのご相談内容
HSPiP用PCの選定にあたり、推奨される構成を知りたい。 ソフトウェアライセンスやインストールは含まず、PC単体での見積もりを希望。 テグシスの過去の構成事例で英語版OSがインストールされている背景や、現在の対応状況についても確認したい。テガラからのご提案
CPUについて HSPiPの処理ではGPUやメモリの要求が低く、解析性能は主にCPUに依存します。 本構成では処理速度と安定性を確保するために、現行世代の高性能CPUであるIntel Core Ultra 9 285 (2025年12月現在) を採用しました。 CPU性能を抑えてコストを重視した構成は PC-TUNS254275でご確認いただけます。 OSの選定について 過去の事例では、日本語版O…主な仕様
CPU Intel Core Ultra 9 285 2.50GHz(8C/8T)+1.90GHz(16C/16T) メモリ 合計8GB DDR5 6400 8GB x 1 ストレージ1 500GB SSD S-ATA ビデオ NVIDIA RTX A400 4GB (MiniDisplayPort x4) ネットワーク on board(2.5GBase-T x1) Wi-Fi 6E,Bluetooth 筐体+電源 ミニタワー型筐体+550W 80PLUS BRONZE OS Microsoft Windows 11 Professional 64bit 英語版
- 事例No.PC-TUNS254275
-
参考価格 (税込):
243,100円事例追加日:2025/12/19HSPiP対応ワークステーション (コスト重視構成)
用途:HSPiPお客さまからのご相談内容
HSPiP用PCの選定にあたり、最低限解析できる構成を知りたい。 ソフトウェアライセンスやインストールは含まず、PC単体での見積もりを希望。 過去の事例で英語版OSがインストールされている背景や、現在の対応状況についても確認したい。テガラからのご提案
コストを抑えつつ、安定した計算を実現するための構成をご提案しました。 CPUについて HSPiPの処理ではGPUやメモリの要求が低く、解析性能は主にCPUに依存します。 本構成では、導入しやすさとコストパフォーマンスを重視し、Intel Core Ultra 5 225を選定しました。 なお、処理速度をさらに確保した構成はPC-TUNS254264でご確認いただけます。 OSの選定について 過去の…主な仕様
CPU Intel Core Ultra 5 225 3.30GHz(6C/6T)+2.70GHz(4C/4T) メモリ 合計8GB DDR5 6400 8GB x 1 ストレージ1 500GB SSD S-ATA ビデオ NVIDIA RTX A400 4GB (MiniDisplayPort x4) ネットワーク on board(2.5GBase-T x1) Wi-Fi 6E,Bluetooth 筐体+電源 ミニタワー型筐体+550W 80PLUS BRONZE OS Microsoft Windows 11 Professional 64bit 英語版
ご注文の流れ
 |
お問い合わせフォームよりご相談内容をお書き添えの上、 お問い合わせください。 (お電話でもご相談を承っております) |
 |
弊社より24時間以内にメールにてご連絡します。 |
 |
必要に応じてメールにて打ち合わせさせていただいた上で、 メール添付にてお見積書をお送りします。 |
 |
お見積もり内容にご納得いただけましたら、メールにてご注文ください。 ご注文確定後、必要な部材を手配し PCを組み立てます。 (掛売りの場合、最初に新規取引票のご記入をお願いしております) |
 |
動作チェックなどを行い、納期が確定いたしましたらご連絡いたします。 (納期は仕様や製造ラインの状況により異なります) |
 |
お客様のお手元にお届けいたします (ヤマト運輸/西濃運輸) |
お支払い方法
お支払い方法は、お見積もりメール・お見積書でもご案内しています。
| 法人掛売りのお客様 |
| 原則として、月末締、翌月末日払いの後払いとなります。 |
| 学校、公共機関、独立行政法人のお客様 |
| 納入と同時に書類三点セット(見積書、納品書、請求書)をお送りしますのでご請求金額を弊社銀行口座へ期日までにお振込み願います。 先に書面での正式見積書(社印、代表者印付)が必要な場合はお知らせください。 |
| 企業のお客様 |
| 納品時に、代表者印つきの正式書類(納品書、請求書)を添付いたします。 ご検収後、請求金額を弊社銀行口座へお支払い期日までにお振込み願います。 |
| 銀行振込(先振込み)のお客様 |
| ご注文のご連絡をいただいた後、お振込みを確認した時点で注文の確定とさせていただきます。 |
修理のご依頼・サポートについて
弊社製PCの保証内容は、お見積もりメールでもご案内しています。
■お問合せ先
テガラの取り扱い製品に関する総合サポート受付のWEBサイトをご用意しております。
テガラ株式会社 サポートサイト
※お問い合わせの際には、「ご購入前」と「ご購入後」で受付フォームが分かれておりますので、ご注意ください。
| メール | support@tegara.com |
| 電話 | 053-543-6688 |
■テグシスのサポートについて
保証期間内の修理について
保証期間内におけるハードウェアの故障や不具合につきましては、無償で修理いたします。
ただし、お客様による破損や、ソフトウェアに起因するトラブルなど保証規定にて定める項目に該当する場合は保証対象外となります。
保証期間経過後も、PCをお預かりしての初期診断は無料で実施しております。
無料メール相談
PCの運用やトラブルにつきまして、メールでのご相談を承ります。経験・知識の豊富な技術コンサルタントが無料でアドバイスいたします。
※調査や検証が必要な場合はお答えできなかったり、有償対応となることがあります
オプション保証サービス
| 「あんしん+」 もしもの時の延長保証サービス |
|
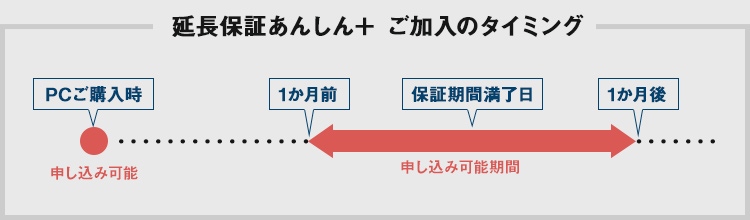
PCのご購入時にトータル5年までの延長保証をご選択いただけます。また、ご購入後にも延長保証を申し込むことができます。
|

| HDD返却不要サービス |
|
保証期間内にPCのHDD(SSD)が故障した場合、通常、新品のHDDとの交換対応となり、故障したHDDはご返却いたしません。 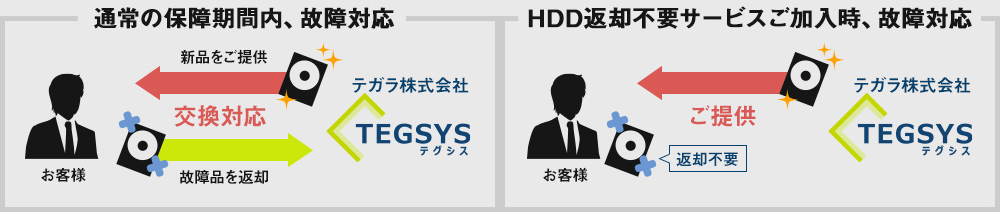
|

| オンサイト保守サポート | |
|
故障発生時、必要に応じエンジニアスタッフが現地へ訪問し、保守対応を行うサービスです。
|
「お客様だけのオーダーメイドPC」を製作しています。
用途に応じた細かなアドバイスや迅速な対応がテガラの強みです。
上記の仕様はテガラでお客様に提案したPC構成の一例です。
掲載内容は提案当時のものであり、また使用する部材の供給状況によっては、現在では提供がむずかしいものや、部材を変更してのご提案となる場合がございます。
参考価格については、提案当時の価格(送料込・税込)になります。
ご相談時期によっては価格が異なる場合がございますので、あらかじめご了承ください。




